森本耕三*,**, 土方美奈子**, Tz-Chun Guo**,宮林亜希子**, 山田博之**, 慶長直人*,**
*公益財団法人結核予防会複十字病院(〒204-8522 東京都清瀬市松山3-1-24)
**公益財団法人結核予防会結核研究所
Primary ciliary dyskinesia
Kozo Morimoto*,**, Minako Hijikata**, Tz-Chun Guo**, Akiko Miyabayashi**, Hiroyuki Yamada**, Naoto Keicho*,**,
*Fukujuji Hospital, Japan Anti-Tuberculosis Association, Tokyo
**The Research Institute of Tuberculosis, Japan Anti-Tuberculosis Association, Tokyo
Keywords:原発性線毛機能不全症候群,鼻腔一酸化窒素濃度測定,遺伝子解析,電子顕微鏡,気管支拡張症/primary ciliary dyskinesia, nasal nitric oxide measurement, genetic analysis, electron microscopy,bronchiectasis
呼吸臨床 2020年4巻6号 論文No.e00103
Jpn Open J Respir Med 2020 Vol. 4 No. 6 Article No.e00103
DOI: 10.24557/kokyurinsho.4.e00103
受付日:2020年5月1日
掲載日:2020年6月22日
©️Kozo Morimoto, et al. 本論文はクリエイティブ・コモンズ・ライセンスに準拠し,CC-BY-SA(原作者のクレジット[氏名,作品タイトルなど]を表示し,改変した場合には元の作品と同じCCライセンス[このライセンス]で公開することを主な条件に,営利目的での二次利用も許可されるCCライセンス)のライセンシングとなります。詳しくはクリエイティブ・コモンズ・ジャパンのサイト(https://creativecommons.jp/)をご覧ください。
PCDはSBSに含まれる。SBSの診断は問診や画像検索などから比較的容易とされるが,一般的にはSBSの原因となる疾患鑑別は行われないことも多いと思われる。さらに,喘息に合併する好酸球性副鼻腔炎症例の混在により臨床所見も複雑化しその理解が進まずにいる。
ATSガイドラインは臨床症状に注目した研究から,PCD症例には①通年性の湿性咳嗽,②出生後からはじまる通年性の鼻副鼻腔炎症状,③約80%に認める出生時呼吸不全,④約50%に認める内臓逆位,が特徴的であり,このうち2つ以上を認める症例に検査(専門施設紹介)を推奨している。
本邦では未診断の成人PCD症例が多数存在することから,出生時の呼吸不全や呼吸器症状出現時期がわからないことが多いと予想されるため,広くSBS症例にPCD検索を行うべきと思われる。また,後述する,マクロライド療法不応のびまん性汎細気管支炎やSBSは特に注目される。
PCD診断のために用いられる主な検査には,鼻腔一酸化窒素濃度測定(nNO測定),EM検査,遺伝子検査,High Speed Video Microscopy(HSVM),免疫蛍光染色Immunofluorescenceがある。単一検査での診断は不可能であり複数の検査の組み合わせにより行われる。欧米では囊胞性線維症がPCDよりも高率に認められるため,その遺伝子検査を先行させ除外することが必要とされる。一方で,本邦では,その多くが未診断の状態であり,まず何よりも疑うことが重要である。また,肺非結核性抗酸菌症症(肺NTM症)が増加しており,類似の細気管支炎や気管支拡張病変などの画像所見を示すPCD患者に肺NTM症を発症した場合には,PCDを疑わなければ肺NTM症のみで診療を受けることになることにも注意が必要である[1][2][3]。
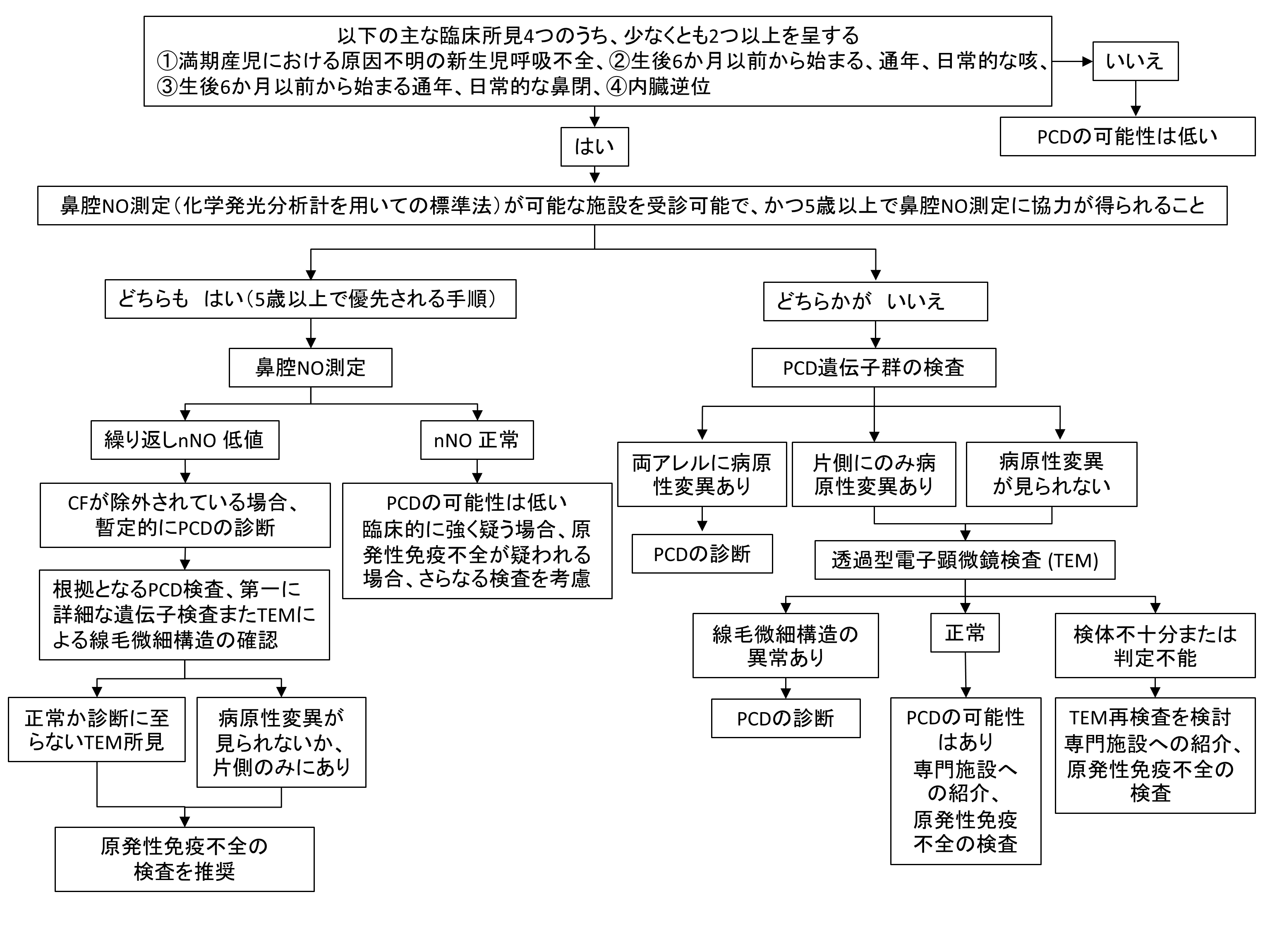
図2 米国胸部疾患学会(ATS)のガイドライン和訳
詳しくは原典を参照。
PCD:原発性線毛機能不全症候群,nNO:鼻腔一酸化窒素濃度測定,CF:囊胞性線維症
(文献[1]より改変引用)
1.鼻腔NO濃度測定[3][4]
PCDでは機序についてはいまだに明らかにはなっていないが,鼻腔一酸化窒素濃度が低値となるため,侵襲も少ないことから最も有用な検査の1つとされている。ATSガイドラインには,臨床所見で疑った症例に第一に行うべき検査として記されており,カットオフ値77nl/minで,感度特異度が96%以上でPCD症例を検出できるとされる。しかし,PCDのなかでも稀ではあるが原因遺伝子(GAS8,RSPH1)によっては正常範囲を示すものもあることが知られており,臨床的に疑わしい症例には正常値であっても精査をすすめる必要がある。われわれの施設では,鼻腔一酸化窒素分析はガイドライン推奨法に従ってレジスター法で行い,Sievers 280i NOA (Sievers,Boulder,CO)により化学発光法で測定している(図3)。呼吸不全を呈し酸素吸入を必要としている低肺機能症例以外には問題なく測定を行えている。低肺機能症例では小児で行うTidal volume法により測定を行っている。

図3 レジスター法を用いた鼻腔一酸化窒素濃度測定
PCDでは低値となる(77nl/minをカットオフとする)。機器によって流量の変更が可能なためppbからnl/minへ変換する。
2.遺伝子検査[5][6]
欧米ではすでに複数の検査会社によって,既知の遺伝子については受託解析が行われており,30種以上の既知のPCD原因遺伝子解析が可能となっている。しかし,検査結果には評価不能な変異を含むこともあり,遺伝子検査だけで簡便に診断が可能というわけではなく30%が未知とされている。評価不能な変異が発見された際にはさらなる検査が必要とされる。現在のところ,日本ではこれらPCDの原因遺伝子セットをターゲットに解析を行う検査会社は知られてない。われわれは,ゲノムDNAからPCRでエクソン領域を増幅し次世代シークエンサー用ライブラリーを作成し,MiSeq(イルミナ)でシークエンスを行う検出法を用い,欧米受託会社と同等の解析を行う体制を確立している(図4,5)。
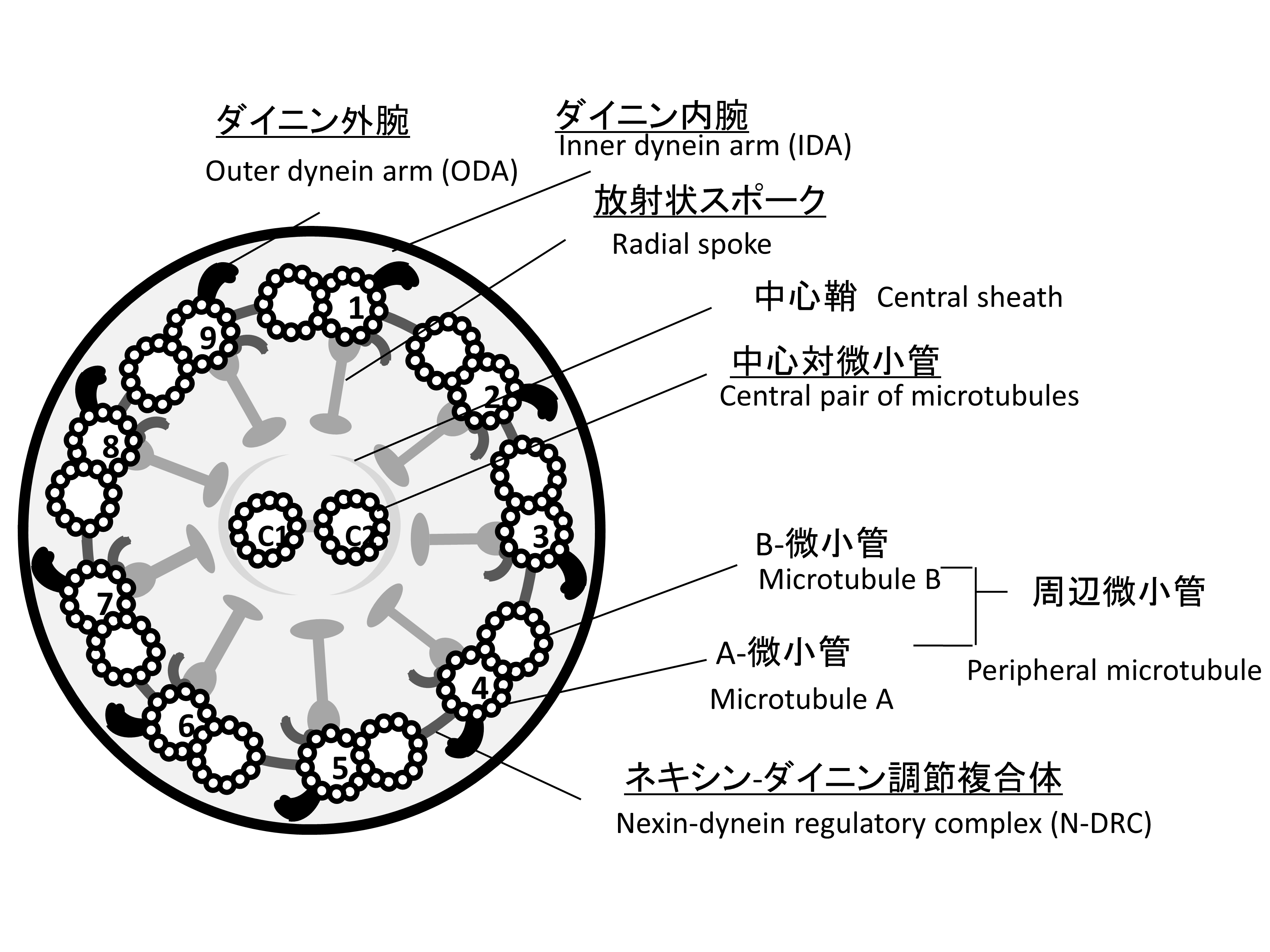
図4 運動性線毛の9+2構造
PCDは主にダイニン腕,Radial Spoke,中心対微小管,ネキシンーダイニン調整複合体などの遺伝子異常によって各部位の構造異常が起こり,線毛の機能不全を来す。
図5 EM所見とPCD原因遺伝子(45遺伝子)
EMでは正常所見をとる遺伝子が複数あることがわかっており,遺伝子検査の重要性が明らかである。一方で,遺伝子検査は,PCDが起こりうると考えられているうちの70%までしかカバーできていないため総合的な診断アプローチを必要とする。結核研究所では2020年4月現在,青字35遺伝子が解析可能。
MTD:microtubular disorganization
3.電子顕微鏡検査(EM)[7]
鼻粘膜,気管支粘膜,精子などから採取された線毛構造異常を検出する。気管支鏡などで用いられる生検鉗子では検体挫滅の問題や異なる細胞の線毛を複数観察することが難しくなるため,鼻粘膜の上皮を専用のキュレットで採取することが一般的である。線毛構造異常としてダイニン腕の欠損が最も多く認められ,欧米ではODA&IDA欠損とODA欠損の頻度が高いとされている(図6)。IDA単独欠損は炎症などによる二次性変化で認められることが多いことから,PCDに特異的な所見ではないことが多いと考えられている。
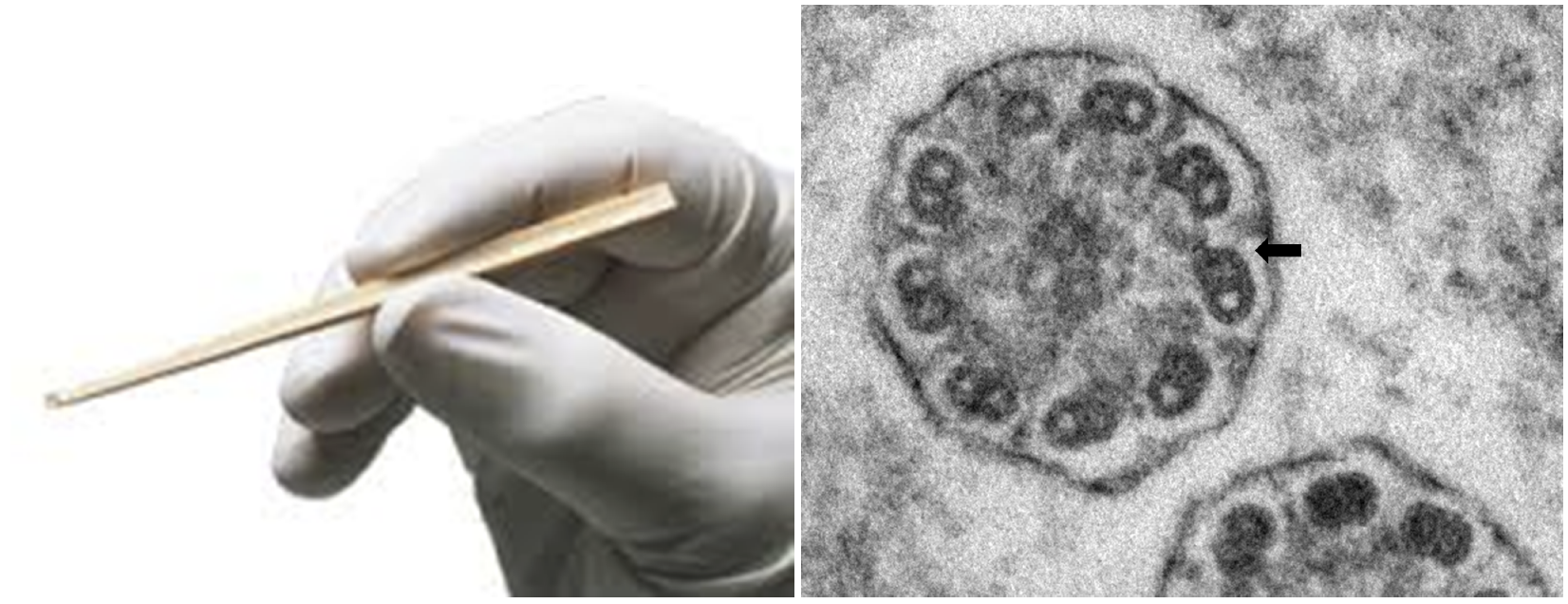
図6 Rhino-Probeによる鼻粘膜生検およびEM所見
Rhino-Probeにより下鼻甲介にcuretteを行う。EM写真は図1に示した症例で,各周辺微小管の外側に見られるべきダイニン外腕(ODA)の欠損を認める。矢印でそのうちの一か所を示す。ARMC4のナンセンス変異を確認した。
4.高速ビデオ顕微鏡分析(high-speed video microscopy:HSVM)
線毛運動のパターンや協調運動障害の有無を判定する。線毛構造が正常でも整列した線毛の協調運動不全を来すこともあり,EM検査では見落とされる症例を発見できる利点があるとされる。欧州呼吸器学会(ERS)ガイドラインではHSVMが推奨に組み込まれているが,熟練した技術者や数回の生検が必要であること,標準化された線毛上皮細胞の培養条件,観察条件がないことなどから,客観的な評価が難しいとする指摘もあり,ATSガイドラインでは推奨されていない。
5.免疫蛍光分析(immunofluorescence)
線毛構造に特異的な蛋白質を蛍光抗体法によって染色し,ダイニン腕や蛋白の欠損を診断する方法。EM検査と異なり,二次性変化による影響を受けないメリットがあるが,主要遺伝子変異に限っても施行可能な施設は少なく,エビデンスとなる十分なデータは今のところ得られていない。
6.サッカリンテスト
偽陽性や偽陰性の確率が高いことから現在では推奨されていない。
7.ATSガイドライン[1][3]
現在,PCDの診断ガイドラインは,ATSとERSによるものが並立している状況にある。両者を比較するとATSではHSVMによる線毛運動の評価を採用しておらず,EM検査よりも遺伝子検査を優先する方針にある。一方,ERSのガイドラインではHSVMは鼻腔一酸化窒素濃度測定とともに主要な検査の1つとなっている。いずれのガイドラインでも確定診断に必要な検査であるEM検査や遺伝子検査,HSVMなどの検査が可能な施設は限られていることから,多くの患者が未診断にある可能性が懸念されている[2]。
これまでの報告の多くは欧米諸国からのものであり,本邦を含む他地域からは症例報告およびケースシリーズにとどまっている。われわれは,1985〜2015年に報告された本邦報告316例のシステマティックレビューを行った。やや男性に多く54.1%(171例),診断時年齢は46.8%が18歳以上で診断され(148/240例),内臓逆位は63%(200/316例)に認めた。PCDでは80%の症例で出生時呼吸不全を呈すると報告されているが,1歳以下の診断は24例(7.6%)のみであった。既往歴には,副鼻腔炎(77.8%,246/316例),気管支拡張症(69.9%,221/316例),繰り返す肺炎(31.3%,99/316例),中耳炎(19.6%,62/316例)と続いた。妊孕性に関する記載のあったうち,男性の85.7%(48/56例),女性56%(14/25例)に不妊があった。
診断時検査は,鼻腔NO測定4例(1.3%),サッカリンテスト33例(10.4%),遺伝子検査5例(1.5%),EM検査230例(72.8%)であり,Kartagenerの3徴候が揃っていれば,それ以上の診断根拠を求めない症例も多く認められた。HSVMやIFを用いた報告はなかった。現行の米国ガイドラインに従って鼻腔NO,EM検査,遺伝子検査を総合的に評価し診断している症例はなかった。また欧米で注目される新生児期呼吸不全についての記載は40例(12.7%)にとどまった。EM所見ではIDA&ODA欠損57例(25.2%),IDA欠損が45例(19.9%)と多く,ODA欠損14例(6.2%)が続き,IDA欠損およびIDA+ODA欠損の割合が高いことが示された。
以上より,欧米の報告に比して診断年齢が高く,ほとんどの症例がEM所見に基づき診断されていること,ODA欠損頻度が低くIDA欠損を根拠にしている場合が多いことが明らかになった。
わが国のPCDの原因遺伝子変異として最も高頻度にみられると推定されるDRC1遺伝子のエクソン1から4までをまたぐ27,748bpの大規模な欠失事例の存在とそのヒトゲノム上の欠失位置を初めて報告した(図7,8)。
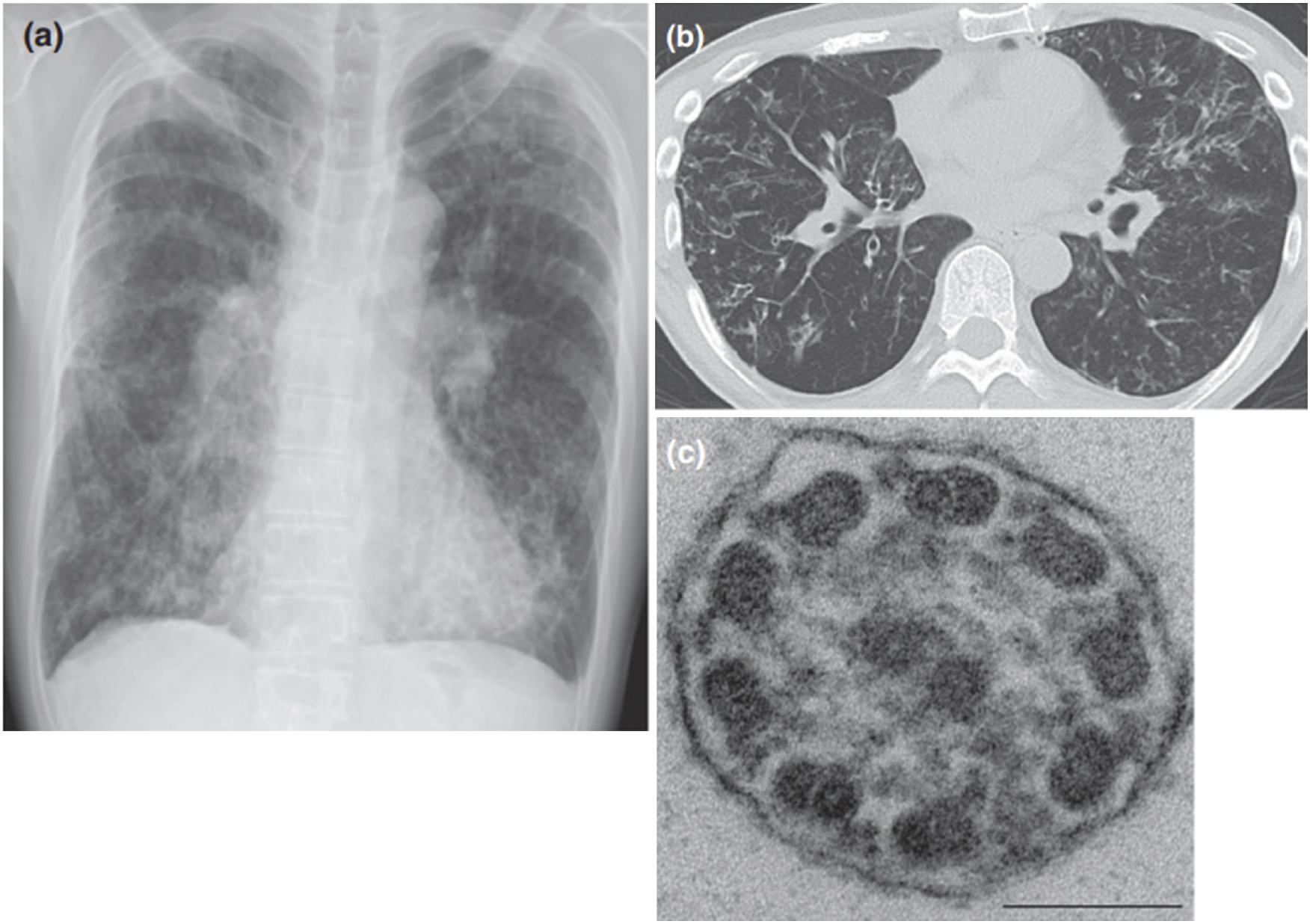
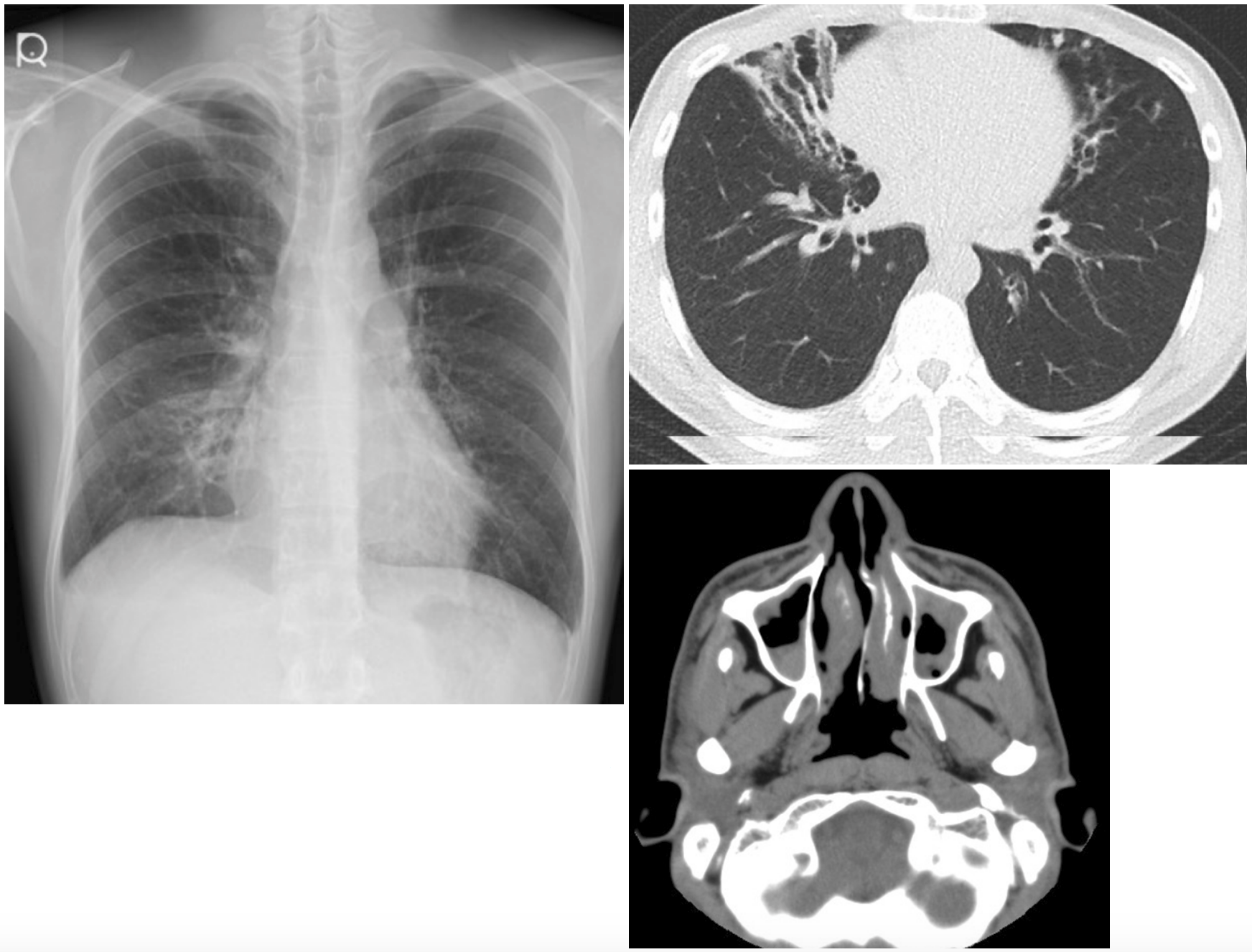
図7 放射線画像(a,b)および鼻粘膜線毛のEM所見(c)
a,b.両側肺びまん性に細葉中心性粒状陰影および気管支拡張を認める。
c.9+2構造が保たれている。(文献[8]より引用)
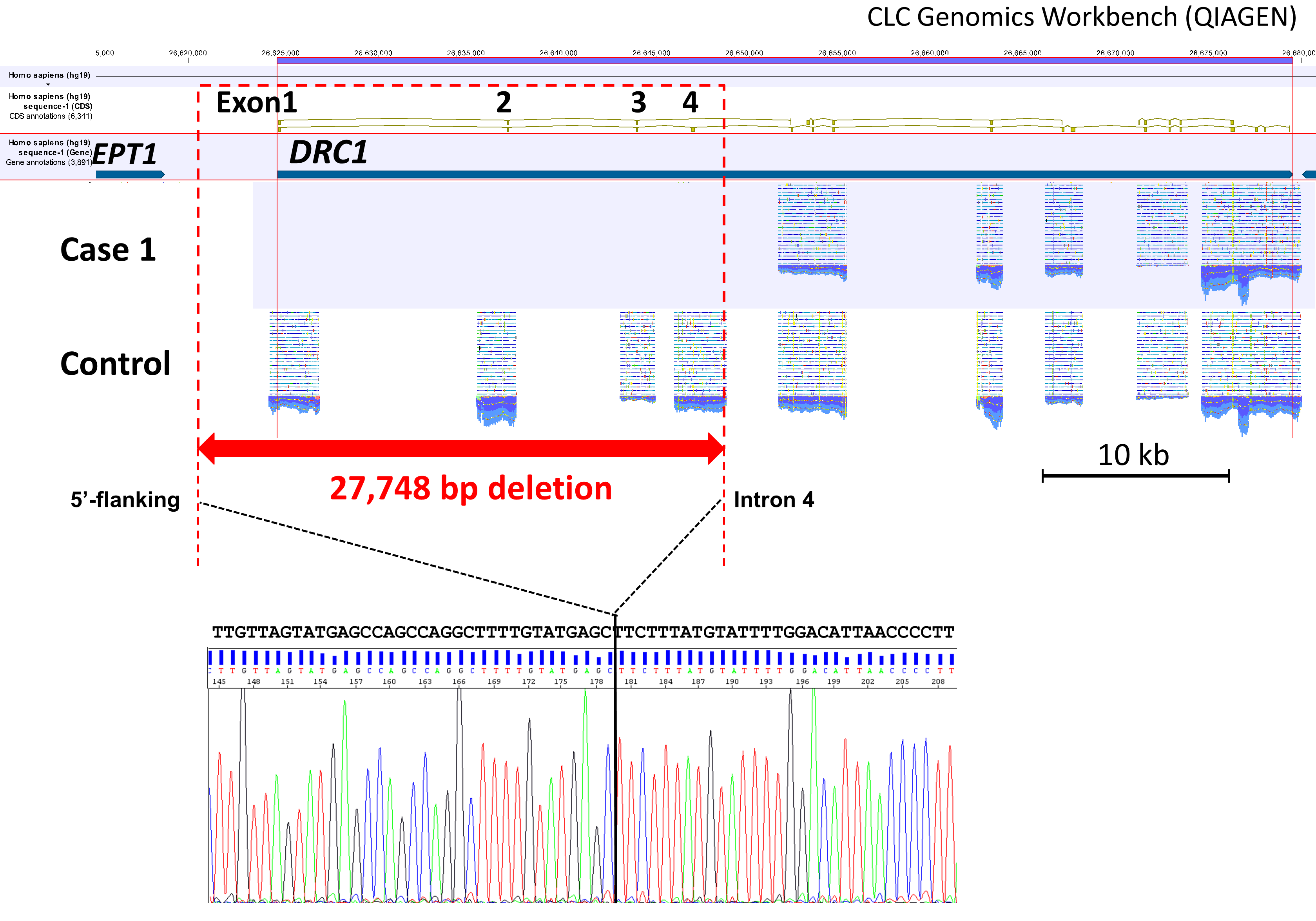
図8 DRC1遺伝子における巨大欠失とそのブレークポイント
PCRと次世代シークエンサー(イルミナ社MiSeq)を用いたターゲットリシークエンスの結果,患者ではDRC1遺伝子のexon1-4にリードがマッピングされず,PCR-サンガーシークエンス法による欠失部位の同定で,27,748bpにわたる巨大欠損をホモまたはヘミ接合で認めた。米国のPCDコホートおよびDNAバンクで同欠失は計5例に同定され,すべてアジア人であった。同欠失はびまん性汎細気管支炎の保存DNAの解析により2%に認めた。
症例は50代男性。40代後半に近医でDPBと診断されマクロライドを投与されるも不応を示し在宅酸素療法が導入された。症状の増悪傾向を訴え当院紹介となった。鼻腔NO濃度は低値(5.3nl/min)を示したが,EM所見は正常であった。このため,線毛構造に大きな異常を示さないPCDの原因遺伝子群に絞った変異検索を行ったところ,上記DRC1の巨大欠失がホモまたはヘミ接合で認められた(両アリルがDRC1の巨大欠失の場合はホモ接合,片側のアリルがDRC1の巨大欠失で,もう片側がアリル欠失の場合はヘミ接合であるが,本症例の遺伝子解析範囲では鑑別ができなかった)。
ノースカロライナ大学のデータベースを調べたところ同異常の患者が1例あり,それは韓国人であった。また米国大手遺伝子検査会社(INVITAE)のデータベースを同様に確認したところ同欠失のキャリアーが4例あり,すべてアジア人であった。さらに,同一のDRC1大規模欠失が,びまん性汎細気管支炎の症例定義に合致する症例の中にも複数例見出されることを後方視的研究により確認した[10]。
DRC1の大規模欠失以外に,CCDC39のナンセンス変異,ARMC4遺伝子のナンセンス変異など病原性変異のホモ接合(またはヘミ接合)例を同定している。
PCDは特異的な臨床像がなく,診断に必要な検査が可能な施設が限られていることから,未診断の症例が多く存在すると推測される。副鼻腔気管支症候群,男性不妊,さらに出生時に呼吸不全を呈する症例などでは,PCDを鑑別疾患に挙げて専門施設への紹介を検討するべきと考えられる。
謝辞:本研究は,厚生労働科学研究費補助金 難治性疾患等政策研究事業「びまん性肺疾患に関する調査研究」研究代表者:本間栄(平成26~平成28年度),稲瀬直彦(平成29~令和元年度)により行われています。
本研究は下記助成を受けたものです。
JSPS科研費JP18K08196,JP20K08532
公益財団法人鈴木謙三記念医科学応用研究財団
倫理規定:本研究は,複十字病院倫理委員会(承認番号16024)および結核研究所倫理委員会(承認番号28-20)の承認を受け,全ての患者に書面同意を得て行っている。
利益相反:なし
Primary ciliary dyskinesia (PCD) is a congenital disease characterized by an autosomal recessive or X-linked disorder; it causes abnormalities of motile cilia in the respiratory tract, other organs and sperm flagella. PCD patients show a variety of clinical presentations such as sino-pulmonary symptoms, infertility, and situs inversus. In particular, bronchiectasis is common and can develop into respiratory failure. More than 40 causative genes have been reported but cover only approximately 70% of the variation in PCD. Therefore, a comprehensive approach, including electron microscopic analysis and measurement of nasal nitric oxide, is recommended by ATS guidelines. A systematic review of Japanese cases revealed that the diagnoses of PCD have been made solely by electron microscopic analysis, and the proportion of abnormal findings is different than the proportion in Western countries.
We opened a PCD clinic in Tokyo according to ATS guidelines in 2017. We report a case of a patient with PCD with a deletion in DRC1 who was diagnosed with refractory diffuse panbronchiolitis, and we found that this genotype might be common in the Asian population. Future research and development of new gene panels are warranted to clarify patient characteristics in Asia.